|
HIGH PERFORMANCE LIQUID CHROMATOGRAPHY - HPLC High performance liquid chromatography is a powerful tool in analysis. This page looks at how it is carried out and shows how it uses the same principles as in thin layer chromatography and column chromatography. | ||
|
Note: It is important to read the introductory page about thin layer chromatography before you continue with this one - particularly the part about how thin layer chromatography works. High performance liquid chromatography works on the same basic principle. HPLC is essentially an adaptation of column chromatography - so it might be a good idea to have a (very quick) look at that as well. Use the BACK button on your browser to return quickly to this page. | ||
|
Carrying out HPLC Introduction High performance liquid chromatography is basically a highly improved form of column chromatography. Instead of a solvent being allowed to drip through a column under gravity, it is forced through under high pressures of up to 400 atmospheres. That makes it much faster. It also allows you to use a very much smaller particle size for the column packing material which gives a much greater surface area for interactions between the stationary phase and the molecules flowing past it. This allows a much better separation of the components of the mixture. The other major improvement over column chromatography concerns the detection methods which can be used. These methods are highly automated and extremely sensitive. The column and the solvent Confusingly, there are two variants in use in HPLC depending on the relative polarity of the solvent and the stationary phase. Normal phase HPLC This is essentially just the same as you will already have read about in thin layer chromatography or column chromatography. Although it is described as "normal", it isn't the most commonly used form of HPLC. The column is filled with tiny silica particles, and the solvent is non-polar - hexane, for example. A typical column has an internal diameter of 4.6 mm (and may be less than that), and a length of 150 to 250 mm. Polar compounds in the mixture being passed through the column will stick longer to the polar silica than non-polar compounds will. The non-polar ones will therefore pass more quickly through the column. Reversed phase HPLC In this case, the column size is the same, but the silica is modified to make it non-polar by attaching long hydrocarbon chains to its surface - typically with either 8 or 18 carbon atoms in them. A polar solvent is used - for example, a mixture of water and an alcohol such as methanol. In this case, there will be a strong attraction between the polar solvent and polar molecules in the mixture being passed through the column. There won't be as much attraction between the hydrocarbon chains attached to the silica (the stationary phase) and the polar molecules in the solution. Polar molecules in the mixture will therefore spend most of their time moving with the solvent. Non-polar compounds in the mixture will tend to form attractions with the hydrocarbon groups because of van der Waals dispersion forces. They will also be less soluble in the solvent because of the need to break hydrogen bonds as they squeeze in between the water or methanol molecules, for example. They therefore spend less time in solution in the solvent and this will slow them down on their way through the column. That means that now it is the polar molecules that will travel through the column more quickly. Reversed phase HPLC is the most commonly used form of HPLC. | ||
|
Note: I have been a bit careful about how I have described the attractions of the non-polar molecules to the surface of the stationary phase. In particular, I have avoided the use of the word "adsorpion". Adsorption is when a molecule sticks to the surface of a solid. Especially if you had small molecules in your mixture, some could get in between the long C18 chains to give what is essentially a solution. You could therefore say that non-polar molecules were more soluble in the hydrocarbon on the surface of the silica than they are in the polar solvent - and so spend more time in this alternative "solvent". Where a solute divides itself between two different solvents because it is more soluble in one than the other, we call it partition. So is this adsorption or partition? You could argue it both ways! Be prepared to find it described as either. | ||
|
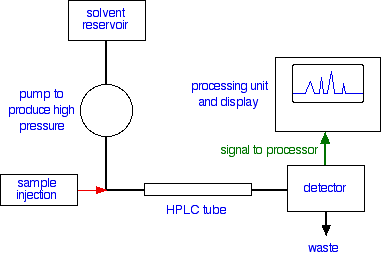
Looking at the whole process A flow scheme for HPLC
Injection of the sample Injection of the sample is entirely automated, and you wouldn't be expected to know how this is done at this introductory level. Because of the pressures involved, it is not the same as in gas chromatography (if you have already studied that). Retention time The time taken for a particular compound to travel through the column to the detector is known as its retention time. This time is measured from the time at which the sample is injected to the point at which the display shows a maximum peak height for that compound. Different compounds have different retention times. For a particular compound, the retention time will vary depending on:
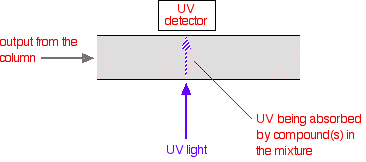
That means that conditions have to be carefully controlled if you are using retention times as a way of identifying compounds. The detector There are several ways of detecting when a substance has passed through the column. A common method which is easy to explain uses ultra-violet absorption. Many organic compounds absorb UV light of various wavelengths. If you have a beam of UV light shining through the stream of liquid coming out of the column, and a UV detector on the opposite side of the stream, you can get a direct reading of how much of the light is absorbed. The amount of light absorbed will depend on the amount of a particular compound that is passing through the beam at the time.
You might wonder why the solvents used don't absorb UV light. They do! But different compounds absorb most strongly in different parts of the UV spectrum. Methanol, for example, absorbs at wavelengths below 205 nm, and water below 190 nm. If you were using a methanol-water mixture as the solvent, you would therefore have to use a wavelength greater than 205 nm to avoid false readings from the solvent. | ||
|
Note: If you are interested, there is a whole section about UV-visible spectroscopy on the site. This explores the question of the absorption of UV and visible light by organic compounds in some detail. | ||
|
Interpreting the output from the detector The output will be recorded as a series of peaks - each one representing a compound in the mixture passing through the detector and absorbing UV light. As long as you were careful to control the conditions on the column, you could use the retention times to help to identify the compounds present - provided, of course, that you (or somebody else) had already measured them for pure samples of the various compounds under those identical conditions. But you can also use the peaks as a way of measuring the quantities of the compounds present. Let's suppose that you are interested in a particular compound, X. If you injected a solution containing a known amount of pure X into the machine, not only could you record its retention time, but you could also relate the amount of X to the peak that was formed. The area under the peak is proportional to the amount of X which has passed the detector, and this area can be calculated automatically by the computer linked to the display. The area it would measure is shown in green in the (very simplified) diagram.
If the solution of X was less concentrated, the area under the peak would be less - although the retention time will still be the same. For example:
This means that it is possible to calibrate the machine so that it can be used to find how much of a substance is present - even in very small quantities. Be careful, though! If you had two different substances in the mixture (X and Y) could you say anything about their relative amounts? Not if you were using UV absorption as your detection method.
In the diagram, the area under the peak for Y is less than that for X. That may be because there is less Y than X, but it could equally well be because Y absorbs UV light at the wavelength you are using less than X does. There might be large quantities of Y present, but if it only absorbed weakly, it would only give a small peak. | ||
|
Note: You will find a useful industry training video which talks through the whole process by following either of these links. The first one has the video embedded in a page of text about the process. The second one has the video on its own on YouTube. Version with written explanation Linking to other sites is always a little bit hazardous because sites change. If you find that this link doesn't work, please contact me via the address on the About this site page. | ||
|
Coupling HPLC to a mass spectrometer This is where it gets really clever! When the detector is showing a peak, some of what is passing through the detector at that time can be diverted to a mass spectrometer. There it will give a fragmentation pattern which can be compared against a computer database of known patterns. That means that the identity of a huge range of compounds can be found without having to know their retention times. | ||
|
Note: If you have forgotten about mass spectrometry, explore the mass spectrometry menu - particularly how a mass spectrometer works, and the formation of fragmentation patterns. | ||
© Jim Clark 2007 (modified June 2016) |
||