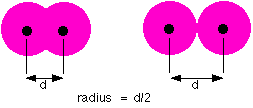|
ATOMIC AND IONIC RADIUS This page explains the various measures of atomic radius, and then looks at the way it varies around the Periodic Table - across periods and down groups. It assumes that you understand electronic structures for simple atoms written in s, p, d notation. | ||||||||||||||||||||||||||||||||||||||||||
|
Important! If you aren't reasonable happy about electronic structures you should follow this link before you go any further. | ||||||||||||||||||||||||||||||||||||||||||
|
ATOMIC RADIUS Measures of atomic radius Unlike a ball, an atom doesn't have a fixed radius. The radius of an atom can only be found by measuring the distance between the nuclei of two touching atoms, and then halving that distance.
As you can see from the diagrams, the same atom could be found to have a different radius depending on what was around it. The left hand diagram shows bonded atoms. The atoms are pulled closely together and so the measured radius is less than if they are just touching. This is what you would get if you had metal atoms in a metallic structure, or atoms covalently bonded to each other. The type of atomic radius being measured here is called the metallic radius or the covalent radius depending on the bonding. The right hand diagram shows what happens if the atoms are just touching. The attractive forces are much less, and the atoms are essentially "unsquashed". This measure of atomic radius is called the van der Waals radius after the weak attractions present in this situation. | ||||||||||||||||||||||||||||||||||||||||||
|
Note: If you want to explore these various types of bonding this link will take you to the bonding menu. | ||||||||||||||||||||||||||||||||||||||||||
|
Trends in atomic radius in the Periodic Table The exact pattern you get depends on which measure of atomic radius you use - but the trends are still valid. The following diagram uses metallic radii for metallic elements, covalent radii for elements that form covalent bonds, and van der Waals radii for those (like the noble gases) which don't form bonds. Trends in atomic radius in Periods 2 and 3
| ||||||||||||||||||||||||||||||||||||||||||
|
Note: You may come across a diagram like this which shows a decreasing trend across a period including the noble gases. You could only get this if you calculated the radius for isolated atoms. It doesn't allow for the real world situation where you actually measure the radius in the way we are discussing here. | ||||||||||||||||||||||||||||||||||||||||||
|
Trends in atomic radius down a group It is fairly obvious that the atoms get bigger as you go down groups. The reason is equally obvious - you are adding extra layers of electrons. Trends in atomic radius across periods You have to ignore the noble gas at the end of each period. Because neon and argon don't form bonds, you can only measure their van der Waals radius - a case where the atom is pretty well "unsquashed". All the other atoms are being measured where their atomic radius is being lessened by strong attractions. You aren't comparing like with like if you include the noble gases.
If you think about it, the metallic or covalent radius is going to be a measure of the distance from the nucleus to the electrons which make up the bond. (Look back to the left-hand side of the first diagram on this page if you aren't sure, and picture the bonding electrons as being half way between the two nuclei.) From lithium to fluorine, those electrons are all in the 2-level, being screened by the 1s2 electrons. The increasing number of protons in the nucleus as you go across the period pulls the electrons in more tightly. The amount of screening is constant for all of these elements. | ||||||||||||||||||||||||||||||||||||||||||
|
Note: You might possibly wonder why you don't get extra screening from the 2s2 electrons in the cases of the elements from boron to fluorine where the bonding involves the p electrons. In each of these cases, before bonding happens, the existing s and p orbitals are reorganised (hybridised) into new orbitals of equal energy. When these atoms are bonded, there aren't any 2s electrons as such. If you don't know about hybridisation, just ignore this comment - you won't need it for UK A level purposes anyway. | ||||||||||||||||||||||||||||||||||||||||||
|
In the period from sodium to chlorine, the same thing happens. The size of the atom is controlled by the 3-level bonding electrons being pulled closer to the nucleus by increasing numbers of protons - in each case, screened by the 1- and 2-level electrons. Trends in the transition elements
Although there is a slight contraction at the beginning of the series, the atoms are all much the same size. The size is determined by the 4s electrons. The pull of the increasing number of protons in the nucleus is more or less offset by the extra screening due to the increasing number of 3d electrons. | ||||||||||||||||||||||||||||||||||||||||||
|
Note: The 4s orbital has a higher energy than the 3d in the transition elements. That means that it is a 4s electron which is lost from the atom when it forms an ion. It also means that the 3d orbitals are slightly closer to the nucleus than the 4s - and so offer some screening. Confusingly, this is inconsistent with what we say when we use the Aufbau Principle to work out the electronic structures of atoms. I have discussed this in detail in the page about the order of filling 3d and 4s orbitals. If you are a teacher or a very confident student then you might like to follow this link. If you aren't so confident, or are coming at this for the first time, I suggest that you ignore it. Remember that the Aufbau Principle (which uses the assumption that the 3d orbitals fill after the 4s) is just a useful way of working out the structures of atoms, but that in real transition metal atoms the 4s is actually the outer, higher energy orbital. | ||||||||||||||||||||||||||||||||||||||||||
|
IONIC RADIUS A warning! Ionic radii are difficult to measure with any degree of certainty, and vary according to the environment of the ion. For example, it matters what the co-ordination of the ion is (how many oppositely charged ions are touching it), and what those ions are. There are several different measures of ionic radii in use, and these all differ from each other by varying amounts. It means that if you are going to make reliable comparisons using ionic radii, they have to come from the same source. What you have to remember is that there are quite big uncertainties in the use of ionic radii, and that trying to explain things in fine detail is made difficult by those uncertainties. What follows will be adequate for UK A level (and its various equivalents), but detailed explanations are too complicated for this level. Trends in ionic radius in the Periodic Table Trends in ionic radius down a group This is the easy bit! As you add extra layers of electrons as you go down a group, the ions are bound to get bigger. The two tables below show this effect in Groups 1 and 7.
| ||||||||||||||||||||||||||||||||||||||||||
|
Note: These figures all come from the Database of Ionic Radii from Imperial College London. I have converted them from Angstroms to nm (nanometres), which are more often used in the data tables that you are likely to come across. If you are interested, 1 Angstrom is 10-10 m; 1 nm = 10-9 m. To convert from Angstroms to nm, you have to divide by 10, so that 1.02 Angstroms becomes 0.102 nm. You may also come across tables listing values in pm (picometres) which are 10-12 m. A value in pm will look like, for example, for chlorine, 181 pm rather than 0.181 nm. Don't worry if you find this confusing. Just use the values you are given in whatever units you are given. For comparison purposes, all the values relate to 6-co-ordinated ions (the same arrangement as in NaCl, for example). CsCl actually crystallises in an 8:8-co-ordinated structure - so you couldn't accurately use these values for CsCl. The 8-co-ordinated ionic radius for Cs is 0.174 nm rather than 0.167 for the 6-co-ordinated version. | ||||||||||||||||||||||||||||||||||||||||||
|
Trends in ionic radius across a period Let's look at the radii of the simple ions formed by elements as you go across Period 3 of the Periodic Table - the elements from Na to Cl.
| ||||||||||||||||||||||||||||||||||||||||||
|
Note: The table misses out silicon which doesn't form a simple ion. The phosphide ion radius is in brackets because it comes from a different data source, and I am not sure whether it is safe to compare it. The values for the sulphide and chloride ions agree in the different source, so it is probably OK. The values are again for 6-co-ordination, although I can't guarantee that for the phosphide figure. | ||||||||||||||||||||||||||||||||||||||||||
|
First of all, notice the big jump in ionic radius as soon as you get into the negative ions. Is this surprising? Not at all - you have just added a whole extra layer of electrons. Notice that, within the series of positive ions, and the series of negative ions, that the ionic radii fall as you go across the period. We need to look at the positive and negative ions separately. The positive ions In each case, the ions have exactly the same electronic structure - they are said to be isoelectronic. However, the number of protons in the nucleus of the ions is increasing. That will tend to pull the electrons more and more towards the centre of the ion - causing the ionic radii to fall. That is pretty obvious! The negative ions Exactly the same thing is happening here, except that you have an extra layer of electrons. What needs commenting on, though is how similar in size the sulphide ion and the chloride ion are. The additional proton here is making hardly any difference. The difference between the size of similar pairs of ions actually gets even smaller as you go down Groups 6 and 7. For example, the Te2- ion is only 0.001 nm bigger than the I- ion. As far as I am aware there is no simple explanation for this - certainly not one which can be used at this level. This is a good illustration of what I said earlier - explaining things involving ionic radii in detail is sometimes very difficult. Trends in ionic radius for some more isoelectronic ions This is only really a variation on what we have just been talking about, but fits negative and positive isoelectronic ions into the same series of results. Remember that isoelectronic ions all have exactly the same electron arrangement.
| ||||||||||||||||||||||||||||||||||||||||||
|
Note: The nitride ion value is in brackets because it came from a different source, and I don't know for certain whether it relates to the same 6-co-ordination as the rest of the ions. This matters. My main source only gave a 4-co-ordinated value for the nitride ion, and that was 0.146 nm. You might also be curious as to how the neutral neon atom fits into this sequence. Its van der Waals radius is 0.154 or 0.160 nm (depending on which source you look the value up in) - bigger than the fluoride ion. You can't really sensibly compare a van der Waals radius with the radius of a bonded atom or ion. | ||||||||||||||||||||||||||||||||||||||||||
|
You can see that as the number of protons in the nucleus of the ion increases, the electrons get pulled in more closely to the nucleus. The radii of the isoelectronic ions therefore fall across this series.
The relative sizes of ions and atoms You probably won't have noticed, but nowhere in what you have read so far has there been any need to talk about the relative sizes of the ions and the atoms they have come from. Neither (as far as I can tell from the syllabuses) do any of the current UK-based exams for 16 - 18 year olds ask for this specifically in their syllabuses. However, it is very common to find statements about the relative sizes of ions and atoms. I am fairly convinced that these statements are faulty, and I would like to attack the problem head-on rather than just ignoring it. Important! For 10 years, until I rewrote this ionic radius section in August 2010, I included what is in the box below. You will find this same information and explanation in all sorts of books and on any number of websites aimed at this level. At least one non-UK A level syllabus has a statement which specifically asks for this.
However, I was challenged by an experienced teacher about the negative ion explanation, and that forced me to think about it carefully for the first time. I am now convinced that the facts and the explanation relating to negative ions are simply illogical. As far as I can tell, no UK-based syllabus mentions the relative sizes of atoms and ions (as of August 2010), but you should check past papers and mark schemes to see whether questions have sneaked in. The rest of this page discusses the problems that I can see, and is really aimed at teachers and others, rather than at students. If you are a student, look carefully at your syllabus, and past exam questions and mark schemes, to find out whether you need to know about this. If you don't need to know about it, stop reading now (unless, of course, you are interested in a bit of controversy!). If you do need to know it, then you will have to learn what is in the box, even if, as I believe, it is wrong. If you like your chemistry to be simple, ignore the rest of the page, because you risk getting confused about what you need to know. If you have expert knowledge of this topic, and can find any flaws in what I am saying, then please contact me via the address on the about this site page. Choosing the right atomic radius to compare with This is at the heart of the problem. The diagrams in the box above, and similar ones that you will find elsewhere, use the metallic radius as the measure of atomic radius for metals, and the covalent radius for non-metals. I want to focus on the non-metals, because that is where the main problem lies. You are, of course, perfectly free to compare the radius of an ion with whatever measure of atomic radius you choose. The problem comes in relating your choice of atomic radius to the "explanation" of the differences. It is perfectly true that negative ions have radii which are significantly bigger than the covalent radius of the atom in question. And the argument then goes that the reason for this is that if you add one or more extra electrons to the atom, inter-electron repulsions cause the atom to expand. Therefore the negative ion is bigger than the atom. This seems to me to be completely inconsistent. If you add one or more extra electrons to the atom, you aren't adding them to a covalently bound atom. You can't simply add electrons to a covalently-bound chlorine atom, for example - chlorine's existing electrons have reorganised themselves into new molecular orbitals which bind the atoms together. In a covalently-bound atom, there is simply no room to add extra electrons. So if you want to use the electron repulsion explanation, the implication is that you are adding the extra electrons to a raw atom with a simple uncombined electron arrangement. In other words, if you were talking about, say, chlorine, you are adding an extra electron to chlorine with a configuration of 2,8,7 - not to covalently bound chlorine atoms in which the arrangement of the electrons has been altered by sharing. That means that the comparison that you ought to be making isn't with the shortened covalent radius, but with the much larger van der Waals radius - the only available measure of the radius of an uncombined atom. So what happens if you make that comparison? Group 7
Group 6
Group 5
As we have already discussed above, measurements of ionic radii are full of uncertainties. That is also true of van der Waals radii. The table uses one particular set of values for comparison purposes. If you use data from different sources, you will find differences in the patterns - including which of the species (ion or atom) is bigger. These ionic radius values are for 6-co-ordinated ions (with a slight question mark over the nitride and phosphide ion figures). But you may remember that I said that ionic radius changes with co-ordination. Nitrogen is a particularly good example of this. 4-co-ordinated nitride ions have a radius of 0.146 nm. In other words if you look at one of the co-ordinations, the nitride ion is bigger than the nitrogen atom; in the other case, it is smaller. Making a general statement that nitride ions are bigger or smaller than nitrogen atoms is impossible. So what is it safe to say about the facts? For most, but not all, negative ions, the radius of the ion is bigger than that of the atom, but the difference is nothing like as great as is shown if you incorrectly compare ionic radii with covalent radii. There are also important exceptions. I can't see how you can make any real generalisations about this, given the uncertainties in the data. And what is it safe to say about the explanation? If there are any additional electron-electron repulsions on adding extra electrons, they must be fairly small. This is particularly shown if you consider some pairs of isoelectronic ions. You would have thought that if repulsion was an important factor, then the radius of, say a sulphide ion, with two negative charges would be significantly larger than a chloride ion with only one. The difference should actually be even more marked, because the sulphide electrons are being held by only 16 protons rather than the 17 in the chlorine case. On this repulsion theory, the sulphide ion shouldn't just be a little bit bigger than a chloride ion - it should be a lot bigger. The same effect is shown with selenide and bromide, and with telluride and iodide ions. In the last case, there is virtually no difference in the sizes of the 2- and 1- ions. So if there is some repulsion playing a part in this, it certainly doesn't look as if it is playing a major part. What about positive ions? Whether you choose to use van der Waals radii or metallic radii as a measure of the atomic radius, for metals the ionic radius is smaller than either, so the problem doesn't exist to the same extent. It is true that the ionic radius of a metal is less than its atomic radius (however vague you are about defining this). The explanation (at least as long as you only consider positive ions from Groups 1, 2 and 3) in terms of losing a complete layer of electrons is also acceptable. Conclusion It seems to me that, for negative ions, it is completely illogical to compare ionic radii with covalent radii if you want to use the electron repulsion explanation. If you compare the ionic radii of negative ions with the van der Waals radii of the atoms they come from, the uncertainties in the data make it very difficult to make any reliable generalisations. The similarity in sizes of pairs of isoelectronic ions from Groups 6 and 7 calls into question how important repulsion is in any explanation. Having spent more than a week working on this, and discussing it with input from some very knowledgable people, I don't think there is any explanation which is simple enough to give to most students at this level. It would seem to me to be better that these ideas about relative sizes of atoms and ions are just dropped. At this level, you can describe and explain simple periodic trends in atomic radii in the way I did further up this page, without even thinking about the relative sizes of the atoms and ions. Personally, I would be more than happy never to think about this again for the rest of my life!
© Jim Clark 2000 (last modified January 2022) |
||||||||||||||||||||||||||||||||||||||||||