|
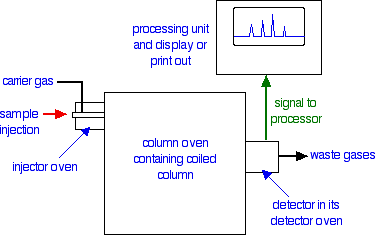
GAS-LIQUID CHROMATOGRAPHY Gas-liquid chromatography (often just called gas chromatography) is a powerful tool in analysis. It has all sorts of variations in the way it is done - if you want full details, a Google search on gas chromatography will give you scary amounts of information if you need it! This page just looks in a simple introductory way at how it can be carried out. Carrying out gas-liquid chromatography Introduction All forms of chromatography involve a stationary phase and a mobile phase. In all the other forms of chromatography you will meet at this level, the mobile phase is a liquid. In gas-liquid chromatography, the mobile phase is a gas such as helium and the stationary phase is a high boiling point liquid adsorbed onto a solid. How fast a particular compound travels through the machine will depend on how much of its time is spent moving with the gas as opposed to being attached to the liquid in some way. A flow scheme for gas-liquid chromatography
| ||
|
Note: You will have to imagine the coiled column in its oven. Drawing a convincing and tidy coil defeated me completely! | ||
|
Injection of the sample Very small quantities of the sample that you are trying to analyse are injected into the machine using a small syringe. The syringe needle passes through a thick rubber disc (known as a septum) which reseals itself again when the syringe is pulled out. The injector is contained in an oven whose temperature can be controlled. It is hot enough so that all the sample boils and is carried into the column as a gas by the helium (or other carrier gas). How the column works The packing material There are two main types of column in gas-liquid chromatography. One of these is a long thin tube packed with the stationary phase; the other is even thinner and has the stationary phase bonded to its inner surface. To keep things simple, we are just going to look at the packed column. The column is typically made of stainless steel and is between 1 and 4 metres long with an internal diameter of up to 4 mm. It is coiled up so that it will fit into a thermostatically controlled oven. The column is packed with finely ground diatomaceous earth, which is a very porous rock. This is coated with a high boiling liquid - typically a waxy polymer. The column temperature The temperature of the column can be varied from about 50°C to 250°C. It is cooler than the injector oven, so that some components of the mixture may condense at the beginning of the column. In some cases, as you will see below, the column starts off at a low temperature and then is made steadily hotter under computer control as the analysis proceeds. How separation works on the column One of three things might happen to a particular molecule in the mixture injected into the column:
None of these things is necessarily permanent. A compound with a boiling point higher than the temperature of the column will obviously tend to condense at the start of the column. However, some of it will evaporate again in the same way that water evaporates on a warm day - even though the temperature is well below 100°C. The chances are that it will then condense again a little further along the column. Similarly, some molecules may dissolve in the liquid stationary phase. Some compounds will be more soluble in the liquid than others. The more soluble ones will spend more of their time absorbed into the stationary phase; the less soluble ones will spend more of their time in the gas. The process where a substance divides itself between two immiscible solvents because it is more soluble in one than the other is known as partition. Now, you might reasonably argue that a gas such as helium can't really be described as a "solvent". But the term partition is still used in gas-liquid chromatography. You can say that a substance partitions itself between the liquid stationary phase and the gas. Any molecule in the substance spends some of its time dissolved in the liquid and some of its time carried along with the gas. Retention time The time taken for a particular compound to travel through the column to the detector is known as its retention time. This time is measured from the time at which the sample is injected to the point at which the display shows a maximum peak height for that compound. Different compounds have different retention times. For a particular compound, the retention time will vary depending on:
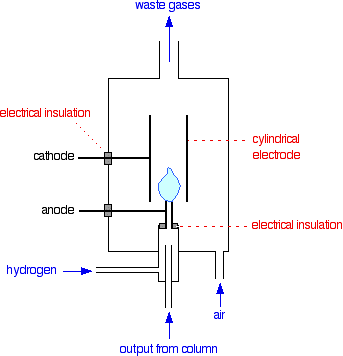
For a given sample and column, there isn't much you can do about the boiling points of the compounds or their solubility in the liquid phase - but you do have control over the temperature. The lower the temperature of the column, the better the separation you will get - but it could take a very long time to get the compounds through which are condensing at the beginning of the column! On the other hand, using a high temperature, everything will pass through the column much more quickly - but less well separated out. If everything passed through in a very short time, there isn't going to be much space between their peaks on the chromatogram. The answer is to start with the column relatively cool, and then gradually and very regularly increase the temperature. At the beginning, compounds which spend most of their time in the gas phase will pass quickly through the column and be detected. Increasing the temperature a bit will encourage the slightly "stickier" compounds through. Increasing the temperature still more will force the very "sticky" molecules off the stationary phase and through the column. The detector There are several different types of detector in use. The flame ionisation detector described below is commonly used and is easier to describe and explain than the alternatives. A flame ionisation detector In terms of reaction mechanisms, the burning of an organic compound is very complicated. During the process, small amounts of ions and electrons are produced in the flame. The presence of these can be detected. The whole detector is enclosed in its own oven which is hotter than the column temperature. That stops anything condensing in the detector.
| ||
|
Note: This is simplified for clarity. There obviously has to be some way of lighting the flame. This is done with an electrically heated coil, but including it clutters the diagram. | ||
|
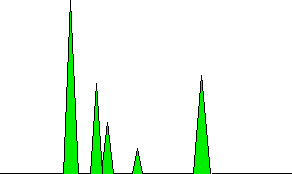
If there is nothing organic coming through from the column, you just have a flame of hydrogen burning in air. Now suppose that one of the compounds in the mixture you are analysing starts to come through. As it burns, it will produce small amounts of ions and electrons in the flame. The positive ions will be attracted to the cylindrical cathode. Negative ions and electrons will be attracted towards the jet itself which is the anode. This is much the same as what happens during normal electrolysis. At the cathode, the positive ions will pick up electrons from the cathode and be neutralised. At the anode, any electrons in the flame will transfer to the positive electrode; and negative ions will give their electrons to the electrode and be neutralised. This loss of electrons from one electrode and gain at the other will result in a flow of electrons in the external circuit from the anode to the cathode. In other words, you get an electric current. The current won't be very big, but it can be amplified. The more of the organic compound there is in the flame, the more ions will be produced, and so the higher the current will be. As a reasonable approximation, especially if you are talking about similar compounds, the current you measure is proportional to the amount of compound in the flame. Disadvantages of the flame ionisation detector The main disadvantage is that it destroys everything coming out of the column as it detects it. If you wanted to send the product to a mass spectrometer, for example, for further analysis, you couldn't use a flame ionisation detector. Interpreting the output from the detector The output will be recorded as a series of peaks - each one representing a compound in the mixture passing through the detector. As long as you were careful to control the conditions on the column, you could use the retention times to help to identify the compounds present - provided, of course, that you (or somebody else) had already measured them for pure samples of the various compounds under those identical conditions. But you can also use the peaks as a way of measuring the relative quantities of the compounds present. This is only accurate if you are analysing mixtures of similar compounds - for example, of similar hydrocarbons. The areas under the peaks are proportional to the amount of each compound which has passed the detector, and these areas can be calculated automatically by the computer linked to the display. The areas it would measure are shown in green in the (very simplified) diagram.
Note that it isn't the peak height that matters, but the total area under the peak. In this particular example, the left-hand peak is both tallest and has the greatest area. That isn't necessarily always so. There might be a lot of one compound present, but it might emerge from the column in relatively small amounts over quite a long time. Measuring the area rather than the peak height allows for this. Coupling a gas chromatogram to a mass spectrometer This can't be done with a flame ionisation detector which destroys everything passing through it. Assuming you are using a non-destructive detector . . . When the detector is showing a peak, some of what is passing through the detector at that time can be diverted to a mass spectrometer. There it will give a fragmentation pattern which can be compared against a computer database of known patterns. That means that the identity of a huge range of compounds can be found without having to know their retention times. | ||
|
Note: If you have forgotten about mass spectrometry, explore the mass spectrometry menu - particularly how a mass spectrometer works, and the formation of fragmentation patterns. | ||
© Jim Clark 2007 (modified July 2016) |
||